What could be the reason for the fact that every time a sample containing SARS and/or FLU virus contigs the meganizing (LR mode) process is stuck? (Even if the max hits parameter is set to 25 in diamond, Megan produces tens of thousands or more of alignments and crashes). Thanks for advance for any idea!
Could you please share the command used along with the corresponding log files for the runs?
Dear Anupam,
Here comes the details (Ubuntu 22.04.3 LTS or Windows 11 OS, -Xmx128000M, 1Tb RAM)
I. LR mode:
- Diamond
diamond blastx -d NR_202306-taxmap -q N152a-103-megahit.fa -o N152a-103-LR --threads 32 -f 100 -c1 -b20 --long-reads
diamond v2.1.9.163 (C) Max Planck Society for the Advancement of Science, Benjamin Buchfink, University of Tuebingen
Documentation, support and updates available at http://www.diamondsearch.org
Please cite: Sensitive protein alignments at tree-of-life scale using DIAMOND | Nature Methods Nature Methods (2021)#CPU threads: 32
Scoring parameters: (Matrix=BLOSUM62 Lambda=0.267 K=0.041 Penalties=11/1)
Temporary directory:
Percentage range of top alignment score to report hits: 10
Opening the database… [0.156s]
Database: NR_202306-taxmap (type: Diamond database, sequences: 542683057, letters: 215028753755)
Block size = 20000000000
Total time = 2177.95s
Reported 7485476 pairwise alignments, 7485476 HSPs.
2897 queries aligned.
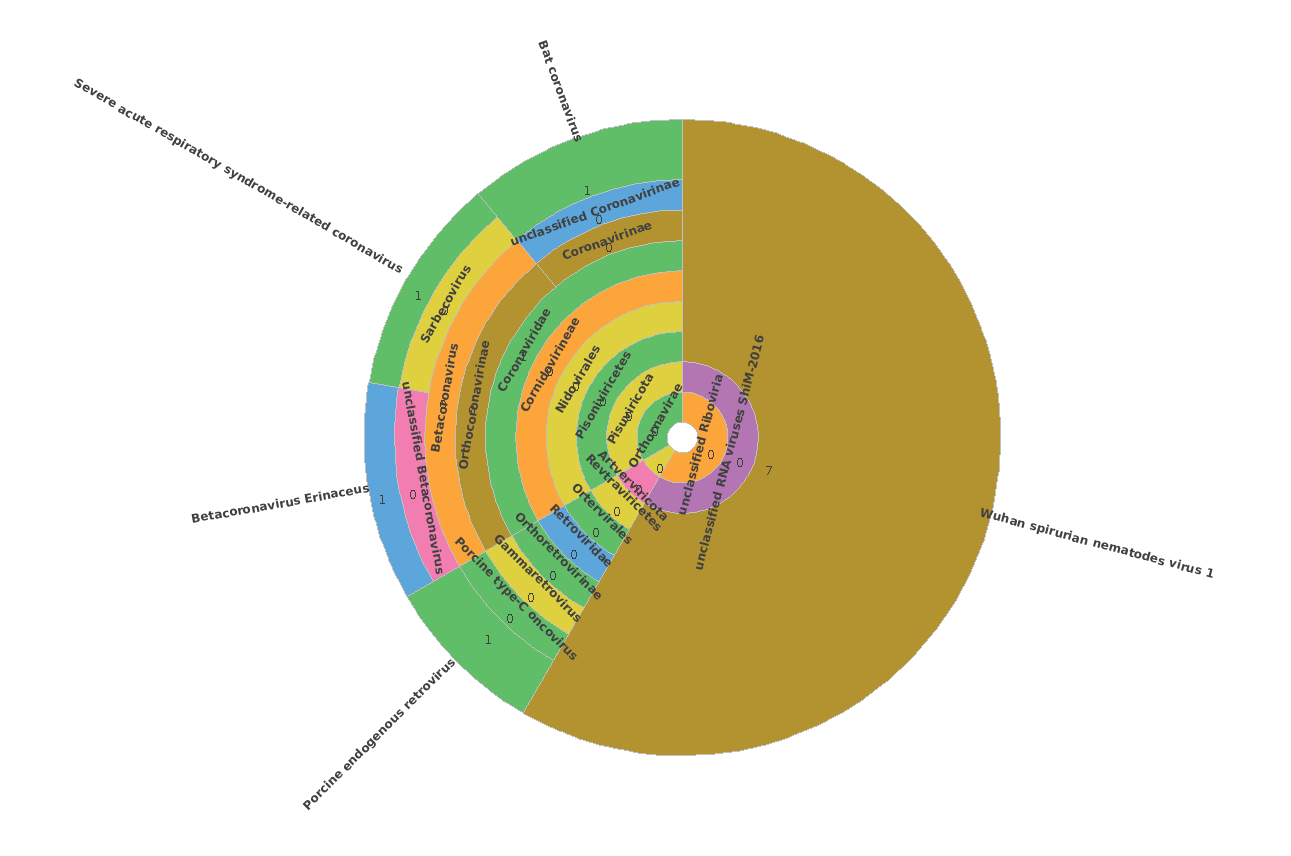
The result is a huge, 1,3Gb sized daa file, with tens of thousands of Coronaviridae aligments. (The maximum number of target sequences per query to report alignments is the default=25).
- Meganizing:
Version MEGAN Community Edition (version 6.25.9, built 16 Jan 2024)
Author(s) Daniel H. Huson
Copyright (C) 2023 Daniel H. Huson. This program comes with ABSOLUTELY NO WARRANTY.
This is free software, licensed under the terms of the GNU General Public License, Version 3.
Sources available at: GitHub - husonlab/megan-ce: MEGAN Community Edition
Java version: 20.0.2; max memory: 125GOpening startup files
Loading ncbi.map: 2,396,736
Loading ncbi.tre: 2,396,740Executing: show window=MeganizeDAA;
Executing: use cViewer=CARD state=false;
Executing: load mapFile=‘/media/ngs_lab/6.4Tb-NVRAM/DB/Megan/megan-map-Feb2022.db’ mapType=MeganMapDB cName=EC;
Executing: use cViewer=EC state=true;
Executing: load mapFile=‘/media/ngs_lab/6.4Tb-NVRAM/DB/Megan/megan-map-Feb2022.db’ mapType=MeganMapDB cName=EGGNOG;
Executing: use cViewer=EGGNOG state=true;
Executing: load mapFile=‘/media/ngs_lab/6.4Tb-NVRAM/DB/Megan/megan-map-Feb2022.db’ mapType=MeganMapDB cName=GTDB;
Executing: use cViewer=GTDB state=true;
Executing: load mapFile=‘/media/ngs_lab/6.4Tb-NVRAM/DB/Megan/megan-map-Feb2022.db’ mapType=MeganMapDB cName=INTERPRO2GO;
Executing: use cViewer=INTERPRO2GO state=true;
Executing: use cViewer=KEGG state=false;
Executing: use cViewer=PGPT state=false;
Executing: load mapFile=‘/media/ngs_lab/6.4Tb-NVRAM/DB/Megan/megan-map-Feb2022.db’ mapType=MeganMapDB cName=SEED;
Executing: use cViewer=SEED state=true;
Executing: load mapFile=‘/media/ngs_lab/6.4Tb-NVRAM/DB/Megan/megan-map-Feb2022.db’ mapType=MeganMapDB cName=Taxonomy;
Executing: use cViewer=Taxonomy state=true;
Executing: update;
Executing: use cViewer=EC state=false;
Executing: use cViewer=EGGNOG state=false;
Executing: use cViewer=GTDB state=false;
Executing: use cViewer=INTERPRO2GO state=false;
Executing: use cViewer=SEED state=false;
Executing: meganize daaFile=‘/home/ngs_lab/diamond/N152a-103-LR.daa’ minScore=50.0 maxExpected=0.01 minPercentIdentity=0.0 topPercent=0.1 minSupport=1 lcaAlgorithm=longReads lcaCoveragePercent=51.0 minReadLength=0 useIdentityFilter=false readAssignmentMode=readCount fNames= longReads=true paired=false;
Meganizing file: /home/ngs_lab/diamond/N152a-103-LR.daa
Annotating DAA file using FAST mode (accession database and first accession per line)
Initializing binning…
Using ‘Interval-Union-LCA’ algorithm (51.0 %) for binning: Taxonomy
Binning reads…
At this step the meganizer always freezes in LR mode, even if there are only 1-2 SARS or FLU contigs in the sample. We have tried about 50-100 samples in the last 2 years, but unfortunately this always happens.
The process also in stucks via command line mode :
daa-meganizer -i /home/ngs_lab/diamond/N152a-103-LR.daa -lg -top 0.1 -supp 0.000000001 -lcp 51 -ram readCount -alg longReads -mdb/home/ngs_lab/DB/Megan/megan-map-Feb2022.db -t 32 -v
II. SR mode without any issue:
- Diamond
diamond blastx -d NR202306-taxmap -q N152a-103.fasta -o N152a-103 --threads 32 -f 100 -c1 -b20
Total time = 4144.54s
Reported 5450167 pairwise alignments, 5450167 HSPs.
344178 queries aligned.
The result is a normal sized (472Mb) daa output.
- Meganizing:
Version MEGAN Community Edition (version 6.25.9, built 16 Jan 2024)
Author(s) Daniel H. Huson
Copyright (C) 2023 Daniel H. Huson. This program comes with ABSOLUTELY NO WARRANTY.
This is free software, licensed under the terms of the GNU General Public License, Version 3.
Sources available at: GitHub - husonlab/megan-ce: MEGAN Community Edition
Java version: 20.0.2; max memory: 125GOpening startup files
Loading ncbi.map: 2,396,736
Loading ncbi.tre: 2,396,740
Executing: show window=MeganizeDAA;
Executing: use cViewer=CARD state=false;
Executing: load mapFile=‘/media/ngs_lab/6.4Tb-NVRAM/DB/Megan/megan-map-Feb2022.db’ mapType=MeganMapDB cName=EC;
Loading ec.map: 8,200
Loading ec.tre: 8,204
Executing: use cViewer=EC state=true;
Executing: load mapFile=‘/media/ngs_lab/6.4Tb-NVRAM/DB/Megan/megan-map-Feb2022.db’ mapType=MeganMapDB cName=EGGNOG;
Loading eggnog.map: 30,875
Loading eggnog.tre: 30,986
Executing: use cViewer=EGGNOG state=true;
Executing: load mapFile=‘/media/ngs_lab/6.4Tb-NVRAM/DB/Megan/megan-map-Feb2022.db’ mapType=MeganMapDB cName=GTDB;
Loading gtdb.map: 240,103
Loading gtdb.tre: 240,107
Executing: use cViewer=GTDB state=true;
Executing: load mapFile=‘/media/ngs_lab/6.4Tb-NVRAM/DB/Megan/megan-map-Feb2022.db’ mapType=MeganMapDB cName=INTERPRO2GO;
Loading interpro2go.map: 14,242
Loading interpro2go.tre: 28,907
Executing: use cViewer=INTERPRO2GO state=true;
Executing: use cViewer=KEGG state=false;
Executing: use cViewer=PGPT state=false;
Executing: load mapFile=‘/media/ngs_lab/6.4Tb-NVRAM/DB/Megan/megan-map-Feb2022.db’ mapType=MeganMapDB cName=SEED;
Loading seed.map: 961
Loading seed.tre: 962
Executing: use cViewer=SEED state=true;
Executing: load mapFile=‘/media/ngs_lab/6.4Tb-NVRAM/DB/Megan/megan-map-Feb2022.db’ mapType=MeganMapDB cName=Taxonomy;
Executing: use cViewer=Taxonomy state=true;
Executing: update;
Executing: use cViewer=EC state=false;
Executing: use cViewer=EGGNOG state=false;
Executing: use cViewer=GTDB state=false;
Executing: use cViewer=INTERPRO2GO state=false;
Executing: use cViewer=SEED state=false;
Executing: meganize daaFile=‘/home/ngs_lab/diamond/N152a-103.daa’ minScore=50.0 maxExpected=0.01 minPercentIdentity=0.0 topPercent=10.0 minSupport=2 lcaAlgorithm=naive minReadLength=0 useIdentityFilter=false readAssignmentMode=readCount fNames= longReads=false paired=false;
Meganizing file: /home/ngs_lab/diamond/N152a-103.daa
Annotating DAA file using FAST mode (accession database and first accession per line)
Initializing binning…
Using ‘Naive LCA’ algorithm for binning: Taxonomy
Binning reads…
Total reads: 344,178
With hits: 344,178
Alignments: 5,450,167
Assig. Taxonomy: 261,104
Min-supp. changes: 2,995
Class. Taxonomy: 5,840
Executing: open file=‘/home/ngs_lab/diamond/N152a-103.daa’;
Info: Opened file ‘N152a-103.daa’ with 344,178 reads
Info: Finished meganizing 1 files.
Info: Command completed (98s): meganize daaFile=‘/home/ngs_lab/diamond/N152a-103.daa’ minScore=50.0 maxExpected=0.01 minPercentIdentity=0.0 topPercent=10.0 …
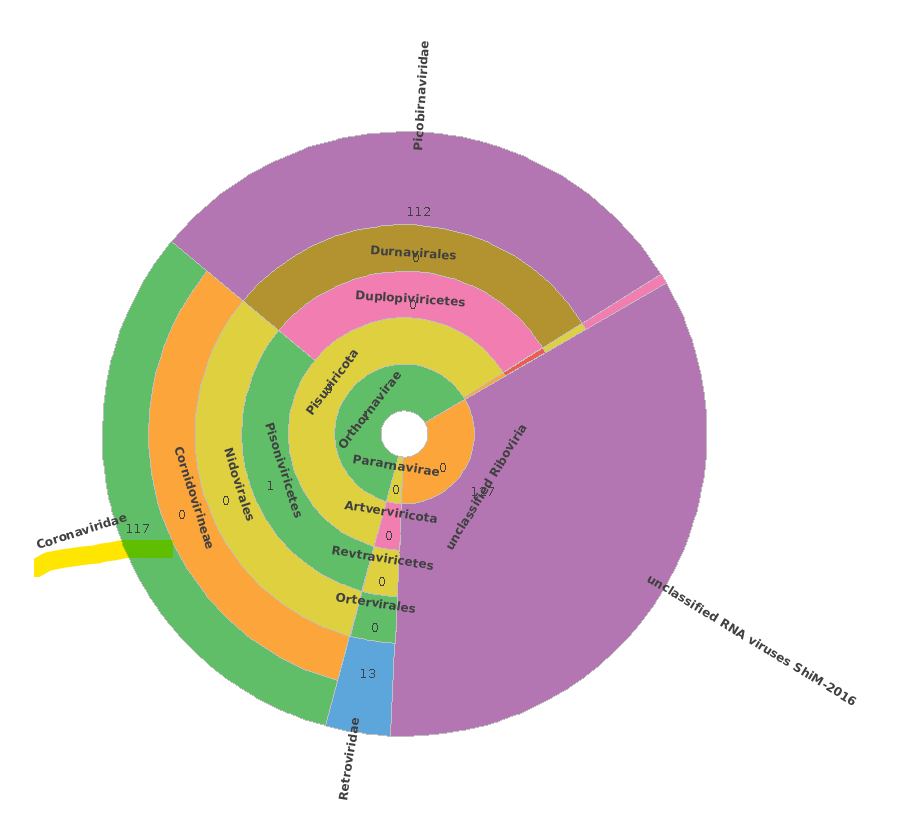
Thank you in advance!
Dear Blaze,
There might not be any issue with these alignments; it’s possible that there are many very similar sequences in the database.
Additionally, the default --long-reads setting will report the top 10 percent of alignments, not the top 25. This is because --long-reads is essentially shorthand for --range-culling --top 10 -F 15, as indicated in the DIAMOND help and your log:
#CPU threads: 32
Scoring parameters: (Matrix=BLOSUM62 Lambda=0.267 K=0.041 Penalties=11/1)
Temporary directory:
Percentage range of top alignment score to report hits: 10
In contrast, with short read mode, you have a k value of 25.
One potential solution for this is to replace --long-reads with -k 25 --range-culling -F 15 to achieve the desired result. However, it’s important to note that the DAA format takes approximately 400 bytes to store one alignment, so the file size may be larger.
Best regards,
Anupam