Dear Daniel and everyone,
How are you doing? I hope you all doing well.
I have a few questions about MEGAN annotation result.
I have fastqc file from 4 soil samples, which is healthy soil and infected soil. I have assembly those file using MEGAHIT and then I run blastx for assembly result using DIAMOND to get .daa file using long reads parameters because I use final contigs for running DIAMOND.
This is my command to run DIAMOND
`
diamond blastx -d nr -q IB_result/final.contigs.fa -o IB_result_LR.daa -f 100 -F 15–range-culling --top 10
`
After I run DIAMOND, then I meganized .daa file using MEGAN6 UE GUI with default LCA parameters and using mapdb file form MEGAN download page.
This is the result for daa meganize
meganize daaFile=‘/home/lulunisrna/Documents/daa-output/daa-output-default-megahit/H1.daa’, '/home/lulunisrna/Documents/daa-…
Initializing binning…
Using ‘Naive LCA’ algorithm for binning: Taxonomy
Using Best-Hit algorithm for binning: SEED
Using Best-Hit algorithm for binning: EGGNOG
Using Best-Hit algorithm for binning: KEGG
Using Best-Hit algorithm for binning: INTERPRO2GO
Binning reads…
Total reads: 1,032,501
With hits: 944,809
Alignments: 2,736,048
Assig. Taxonomy: 931,518
Assig. SEED: 370,058
Assig. EGGNOG: 629,291
Assig. KEGG: 470,047
Assig. INTERPRO2GO: 573,876
Min-supp. changes: 2
Class. Taxonomy: 2,619
Class. SEED: 3,946
Class. EGGNOG: 5,955
Class. KEGG: 6,012
Class. INTERPRO2GO: 5,554
Loading MEGAN File: H1.daa
Meganizing file: /home/lulunisrna/Documents/daa-output/daa-output-default-megahit/H2.daa
Annotating DAA file using FAST mode (accession database and first accession per line)
Initializing binning…
Using ‘Naive LCA’ algorithm for binning: Taxonomy
Using Best-Hit algorithm for binning: SEED
Using Best-Hit algorithm for binning: EGGNOG
Using Best-Hit algorithm for binning: KEGG
Using Best-Hit algorithm for binning: INTERPRO2GO
Binning reads…
Total reads: 764,656
With hits: 698,468
Alignments: 2,055,986
Assig. Taxonomy: 687,687
Assig. SEED: 275,253
Assig. EGGNOG: 460,697
Assig. KEGG: 350,998
Assig. INTERPRO2GO: 429,323
Min-supp. changes: 3
Class. Taxonomy: 2,370
Class. SEED: 3,811
Class. EGGNOG: 5,602
Class. KEGG: 5,733
Class. INTERPRO2GO: 5,360
Loading MEGAN File: H2.daa
Meganizing file: /home/lulunisrna/Documents/daa-output/daa-output-default-megahit/I1.daa
Annotating DAA file using FAST mode (accession database and first accession per line)
Initializing binning…
Using ‘Naive LCA’ algorithm for binning: Taxonomy
Using Best-Hit algorithm for binning: SEED
Using Best-Hit algorithm for binning: EGGNOG
Using Best-Hit algorithm for binning: KEGG
Using Best-Hit algorithm for binning: INTERPRO2GO
Binning reads…
Total reads: 1,244,218
With hits: 1,139,309
Alignments: 3,174,510
Assig. Taxonomy: 1,123,351
Assig. SEED: 435,640
Assig. EGGNOG: 753,171
Assig. KEGG: 553,917
Assig. INTERPRO2GO: 685,877
Min-supp. changes: 3
Class. Taxonomy: 2,675
Class. SEED: 4,099
Class. EGGNOG: 6,335
Class. KEGG: 6,242
Class. INTERPRO2GO: 5,741
Loading MEGAN File: I1.daa
Meganizing file: /home/lulunisrna/Documents/daa-output/daa-output-default-megahit/I2.daa
Annotating DAA file using FAST mode (accession database and first accession per line)
Initializing binning…
Using ‘Naive LCA’ algorithm for binning: Taxonomy
Using Best-Hit algorithm for binning: SEED
Using Best-Hit algorithm for binning: EGGNOG
Using Best-Hit algorithm for binning: KEGG
Using Best-Hit algorithm for binning: INTERPRO2GO
Binning reads…
Total reads: 1,347,086
With hits: 1,228,100
Alignments: 3,530,915
Assig. Taxonomy: 1,210,529
Assig. SEED: 482,355
Assig. EGGNOG: 812,407
Assig. KEGG: 610,620
Assig. INTERPRO2GO: 745,263
Min-supp. changes: 3
Class. Taxonomy: 2,782
Class. SEED: 4,244
Class. EGGNOG: 6,797
Class. KEGG: 6,542
Class. INTERPRO2GO: 5,960
Loading MEGAN File: I2.daa
Info: Finished meganizing 4 files.
Now, I got result for the annotation but the result seems not good because the abundance between healthy and infected soils is quite similar and it’s looks not possible in natural condition.
So, I wonder which one of my step is wrong and how to solve this problem?
What means of number of reads in y axis? is that based on relative abundance or copy number?
Here is one of example of my annotation result for phylum taxonomic of bacteria
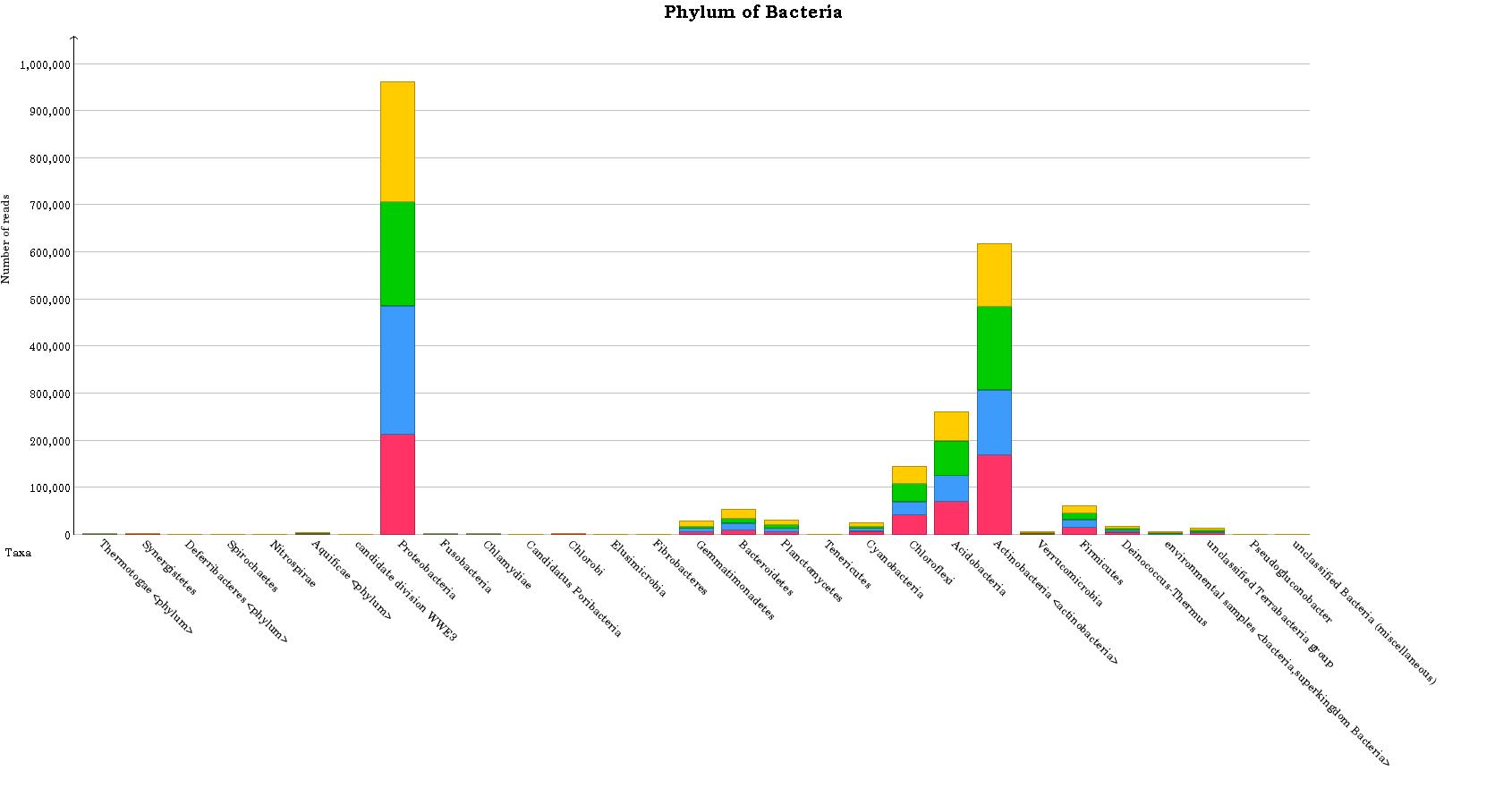
I also got KEGG functional annotation and it looks no connected between taxonomic abundance and functional abundance.
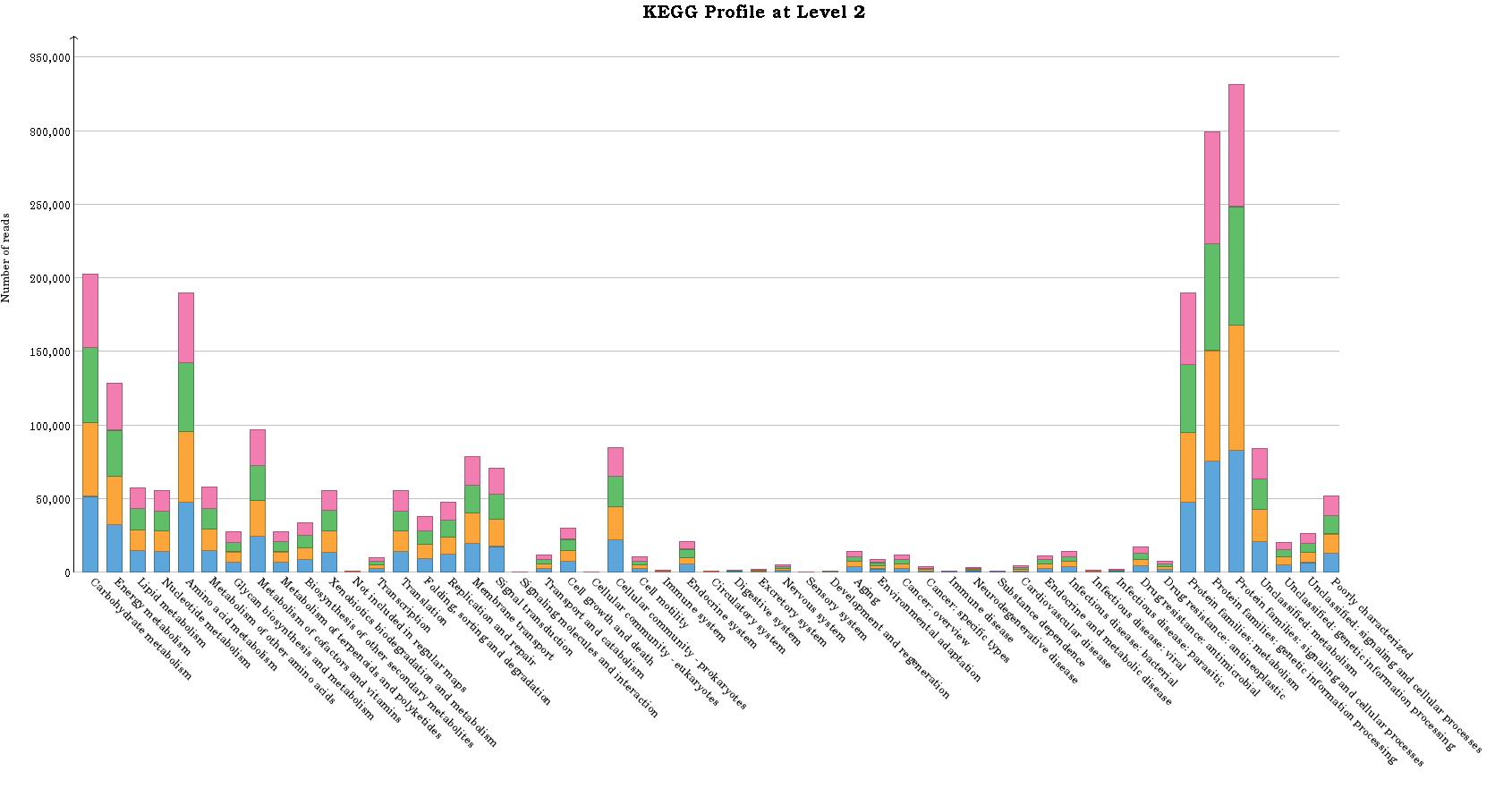
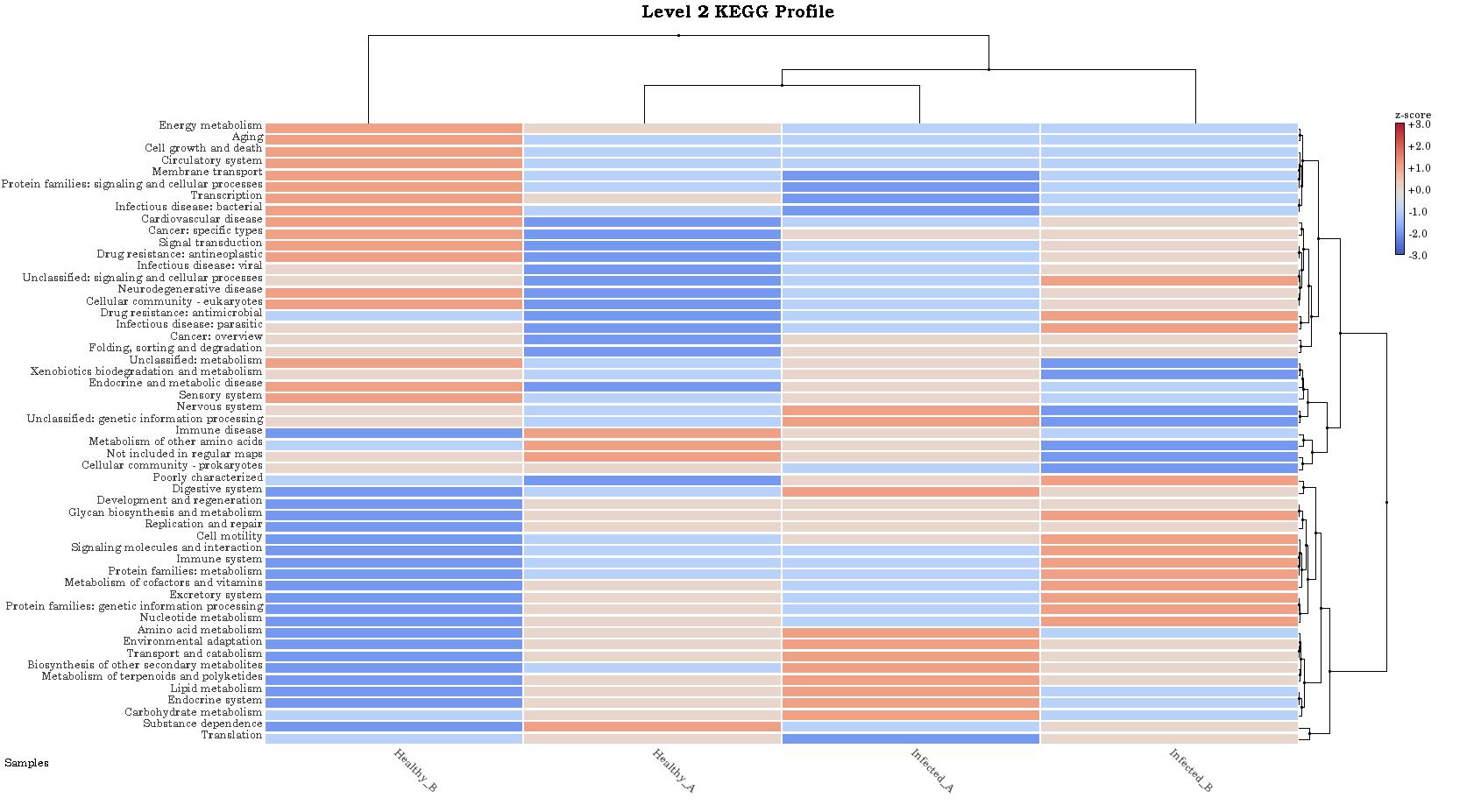
Actually I wonder how MEGAN6 annotate KEGG. Is that relative or linear with taxonomic abudance or how?
Please kindly to give me a guide for this problem.
Thank you very much.
Regards,
Lulu