I have tried running a diamond/megan pipeline on a sample of my data. I used head -n400000 to collect a sample of 100,000 reads from the top of my fastq files (once from each file for paired reads). I aligned these to nr with diamond:
When I import the rma6 file into megan, the total number of reads is shown as 125,752. My question is how is this figure derived? I aligned 100,000 paired reads. Is this considered 200,000 reads by mega, and the difference due to reads not aligned by diamond? For functional/taxonomic quantification, surely paired reads cannot be considered independent counts from one another and therefore a function/taxon should be agreed between a pair to assign a function/taxon for the pair?
I would like to use a feature mentioned in the megan manual: taxon disabling. The manual mentions this is in the options menu and can allow given taxa to be excluded from the analysis, however I can’t see this in the options menu in either the community or ultimate editions of megan6
The fact that MEGAN reports 125,752 reads doesn’t make any sense…
What number of reads did DIAMOND report?
Taxon disabling is in Edit->Preferences->Taxon Disabling…
I would recommend that you use daa-meganizer rather than daa2rma.
This processes your daa file so that it can be opened in megan, is faster and has the advantage of not creating an additional file.
125752=62169 + 63583, so yes, MEGAN counts each read as a single read, and not pairs of reads…
Taxon disabling does not affect functional analysis.
By default, MEGAN will treat each read of a pair of reads as a separate entity. The paired read mode uses some heuristics to try to improve the taxonomic placement of reads that occur in pairs, such as placing read B on the same taxon as read A, if A has a taxonomic assignment and B doesn’t. But I don’t think that the heuristics work well enough to justify the additional hassle of using paired mode.
You can always re-meganize a DAA file if you want to change the analysis, or can also make multiple copies of your DAA file if you want to compare different analyses of the same file side-by-side.
I am pretty sure that a meganized DAA file will be smaller than the corresponding rma6 file. So, the advantages are size and speed (meganizing a DAA file is much faster than creating an rma6 file…)
Perhaps taxon disabling mapping onto the functional analysis would be a useful feature? E.g. this would allow removal of unwanted e.g. host reads and functional analysis without the need for additional steps.
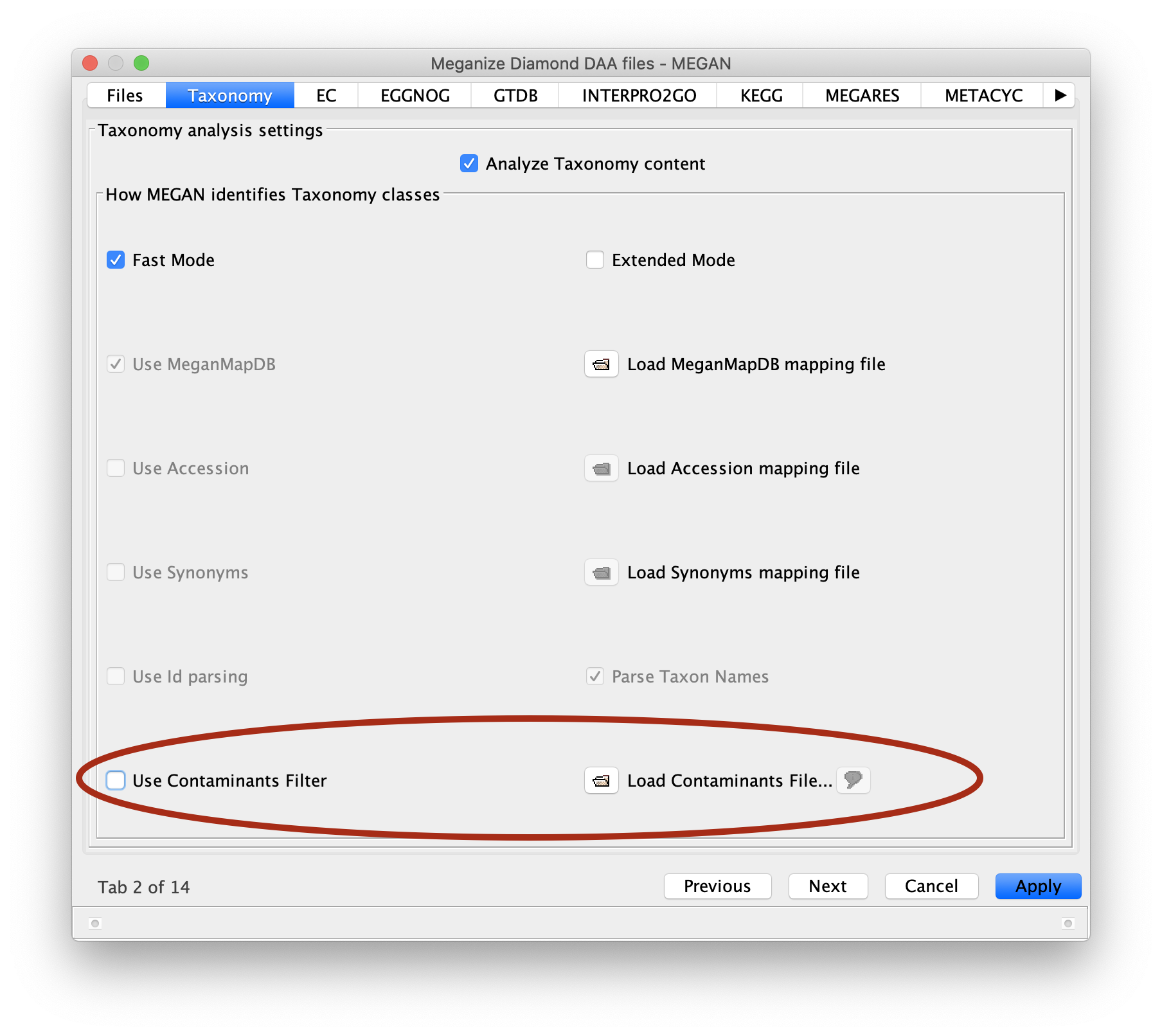
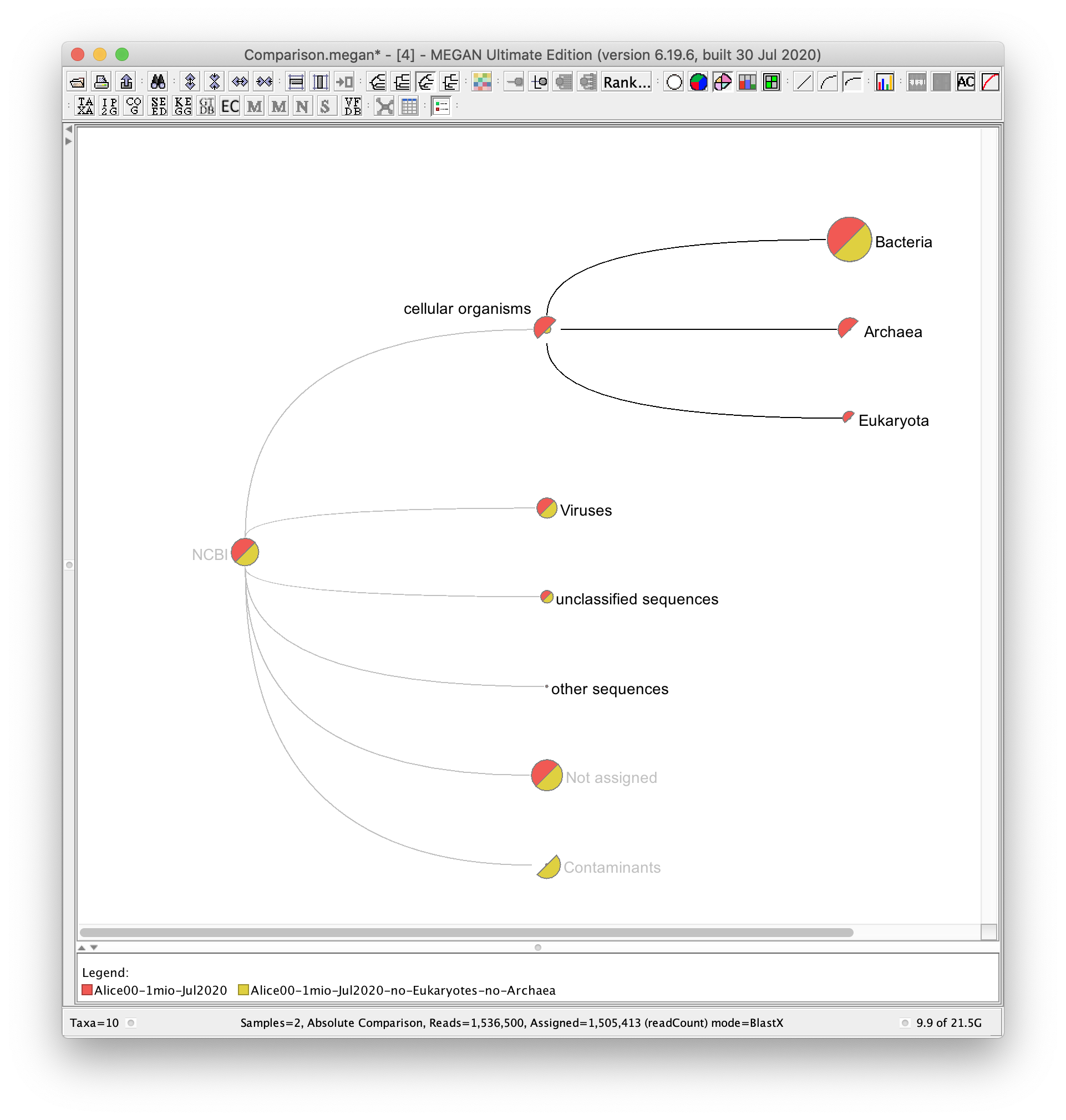
I have implemented contaminant filtering, which is the correct way to address the problem that you mentioned. Any read that aligns to a contaminant is placed on a special contaminant node and is not assigned to any taxon or any function.
Is is quite experimental, so I will need to check that is in working order and then upload a new release in which the feature is activated…
I’d be happy to do so, if you can then give me feed back on whether it does what you desire…
Happy to give this feature a test. I’m interested in how “contaminant” would be defined. Would this be a user-defined set of taxids, as I think the definition of contaminant will vary widely between experiments.
You specify a list of contaminants. For example, if you considered all archaea and eukaryotes to be contaminants, then you would supply the taxon ids 2157 and 2759 during processing of your input file.
MEGAN will report this:
Using contaminants profile: 2 input, 1,522,418 total
This means that you supplied two high-level taxon ids, which sit above a total of 1.5 million ids.
For short reads, any read that has any alignment to some contaminant taxon is put on the contaminant node (where only those alignments are considered that pass all the filters such as top percent, min bitscore etc).
In long read mode, a simpler criterion is used: any read assigned to a contaminant taxon is deemed a contaminant read.
To activate this feature, you must supply a file containing contaminant ids or names, when supplying names, it should be one per line or in single quotes. You will find these at bottom of the Taxonomy tab of the Meganize and Import Blast dialogs.