help.doc (620.5 KB)
You need to provide alignments to MEGAN, not target sequences. See this tutorial for details: Tutorial · husonlab/tutorials Wiki · GitHub
Thanks! To begin the DIAMOND + MEGAN pipeline, the first step is to generate an index for the database file using DIAMOND. The DIAMOND has been installed by using brew (brew install diamond ) on my MacOS. However,for the Index generation, when typing “diamond makedb --in tutorial-nr.gz --db tutorial-nr” , the results will be :
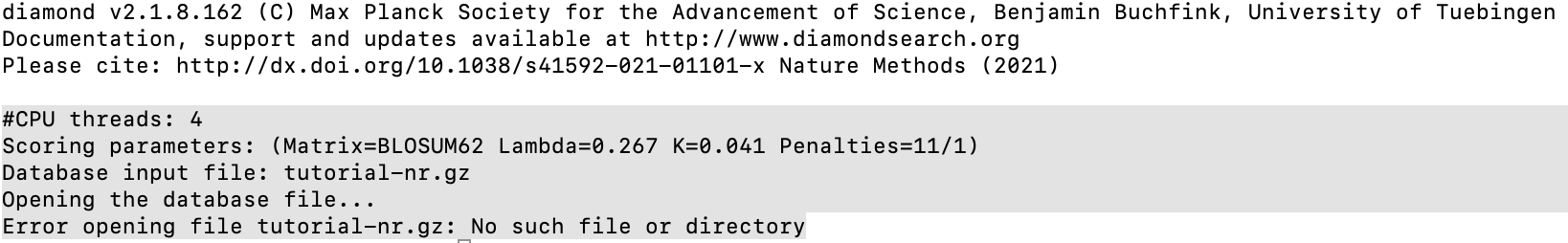
did you download the data file and unzip it? Are you in the directory containing the file tutorial-nr.gz when you typed the command?
To verify, type
ls tutorial-nr.gz
If nothing is listed, the file is not present or you are in the wrong directory.
Please note that the tutorial does assume that you have some command-line knowledge. If you do not know how to create a directory, change directory, list files etc in Linux then please first work through a tutorial that teaches that because unfortunately, our tutorial does not provide that support.
Thank you, Daniel! I still have one question, that is, the total nr database from here: ftp://ftp.ncbi.nih.gov/blast/db/FASTA/nr.gz is too large, could you supply a website only to download the bacteria nr database?
We are working on a clustered version, nr50, that is only 1/10 the size of full NCBI-nr and using this computations run unto 34 times faster
Now , the database nr has been constructed by using the full NCBI-NR, and a protein sequence Acin.fasta was downloaded from the NCBI, how to do the alignment by using diamond? diamond blastp --db nr -q Acin.fasta -o out/Acin.daa -f 100 --masking 0?
Yes, that looks ok, but you probably don’t need this option: --masking 0