I’m blasting virus ASVs. at the moment they are all a single species.
my megan input is a default XML blast output. I get a single node for the virus species.
but there’s additional info in the actual blast match name (strain number for example).
that will be useful to look into diversity.
is there a way to get megan to export the actual blast match name. or their accession number.
I understand this could mean that a single read will contribute to multiple names/numbers.
At the moment, having just taxon name and taxonid is too restrictive for my dataset.
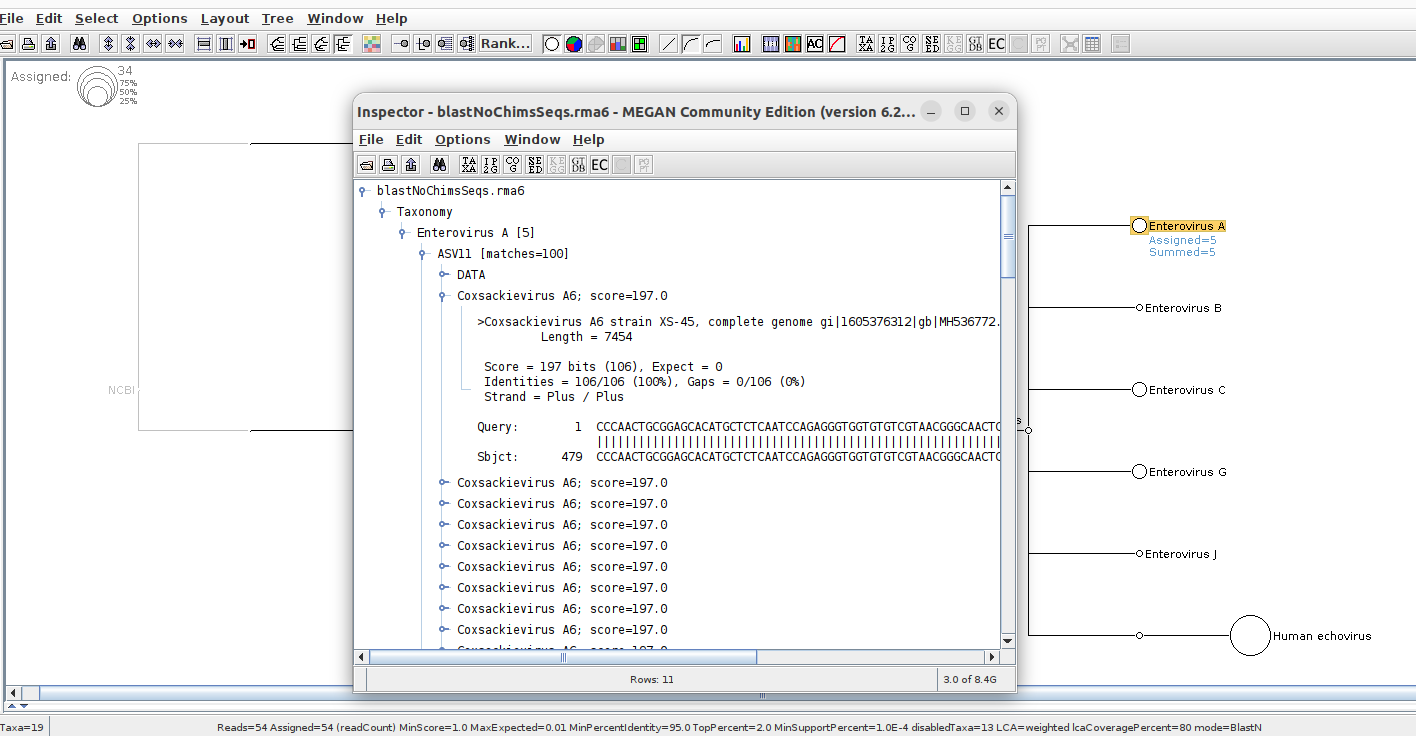
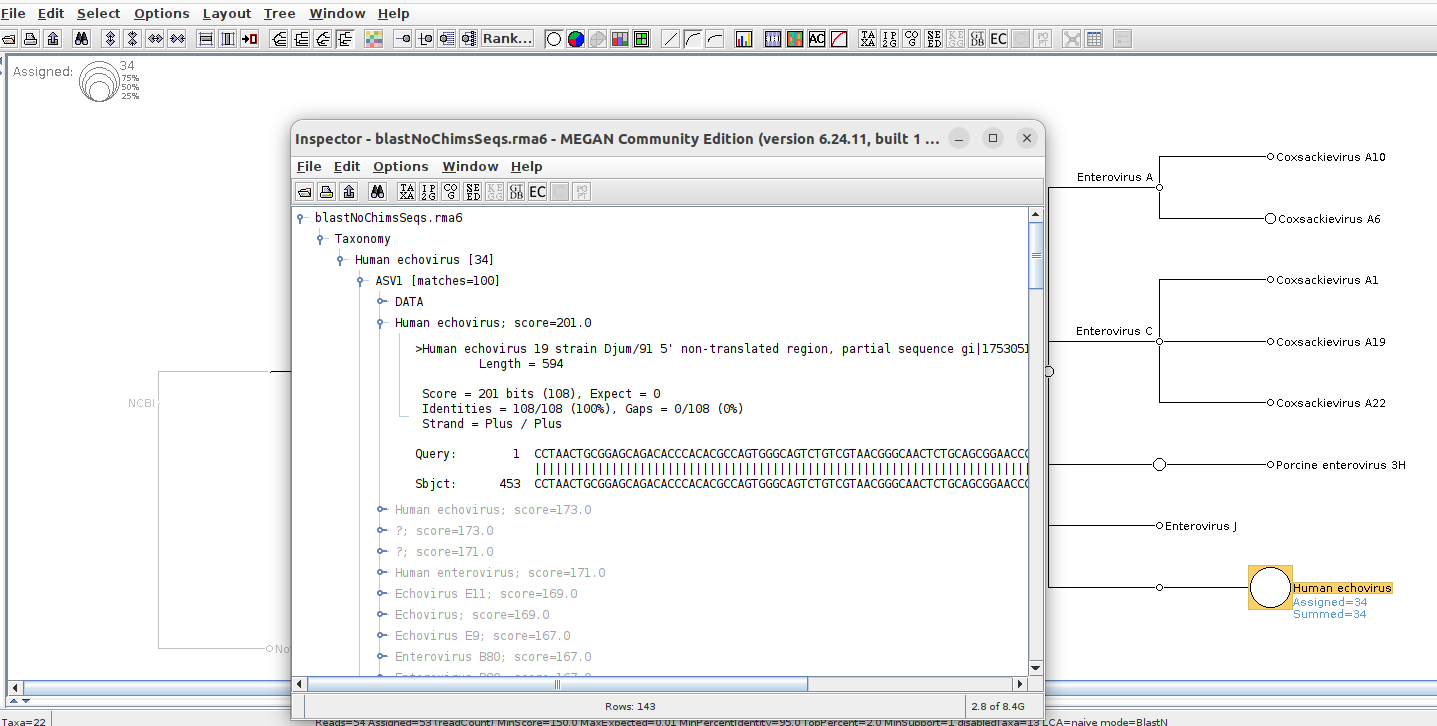
youll see from XML (and inspecting reads in megan) that one of the matches is “Human echovirus 19 strain Djum/91 5' non-translated region, partial sequence”. Megan only displays “Human Echovirus” even in exports. Ideally id want at least “Human echovirus 19 strain Djum/91” .
alternatively, if megan can export the accession numbers for the top hits, I can parse that info elsewhere probably.
Here’s a screenshot of what im looking for.
Many reads get blast-matched to higher level virus levels, but perhaps megan only displays to ‘species’ level. Even exporting will not give me the full match names, (e.g., id like to show that this sample is assigned to Coxsackievirus A6.)
ah it seems i managed to get Coxsackievirus to show up by increasing the bitscore filter.
however, my original question (getting more details on Human Echovirus), remains.
Do you know how to export all that information (posted above) to a .csv file? I want to export all my alignment information (family, genus, specie) with bit score, identities, e-values, length. What can I choose in the .csv export dropdown menu to get this information? I don’t need the alignment sequences. Thanks! @Daniel
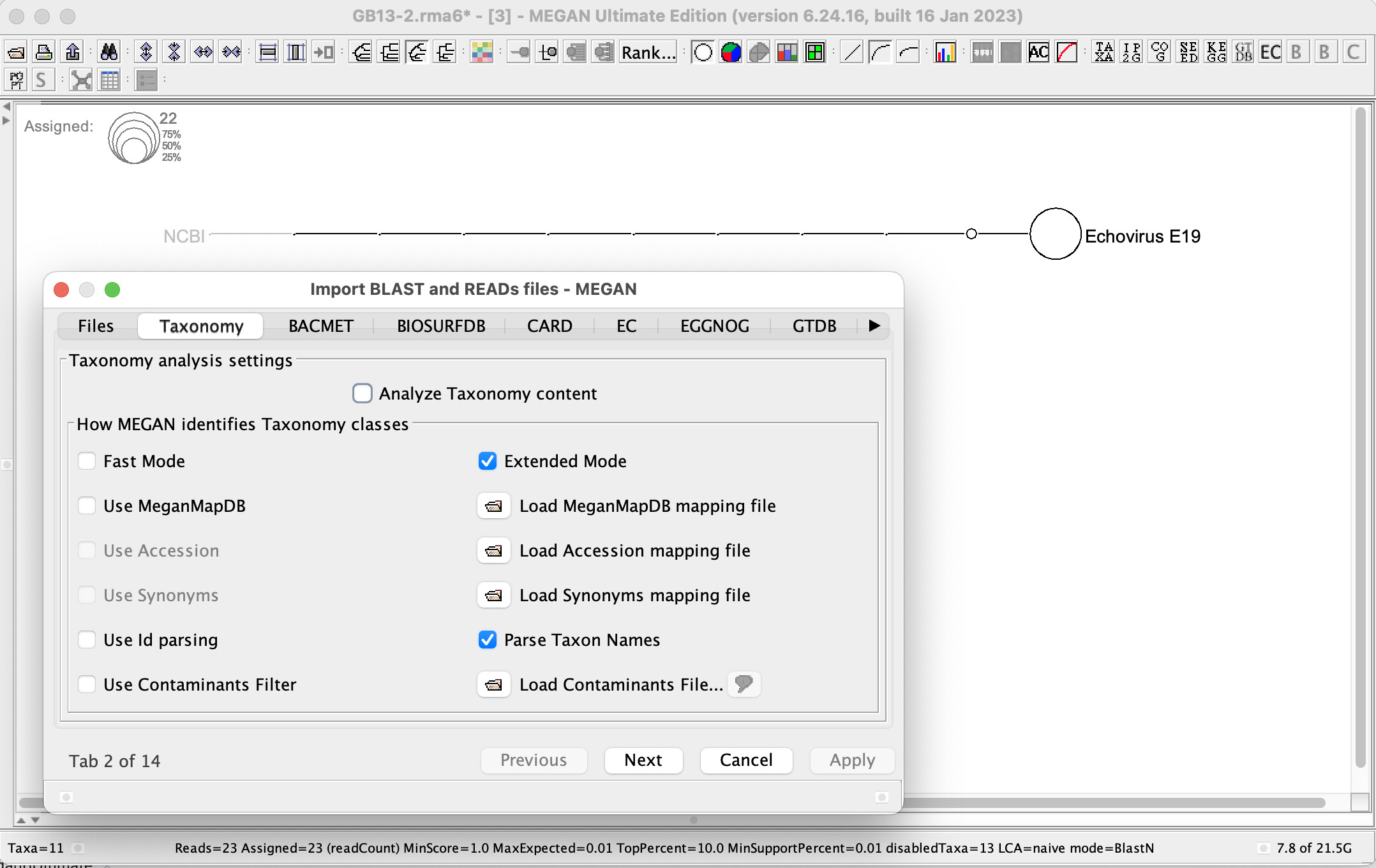
I have looked into this. First, the NCBI taxonomy only goes down Echovirus E19, there are no nodes below that node in the taxonomy. Using Extended Mode and Parse Taxon Names will produce a classification down to the level:
I actually like the way the readName_to_taxonMatches tab separates in Excel. I just with this export gave me the e-value, NCBI accession number, protein name, and assignment name. I am trying to put together a table for my paper that has all this information.
revisiting the same process with a new viral dataset!
previously ive just been using blast.xml files as input into megan.
but i seem to get better resolution when i include a meganmap file. i think the megan-nucl one.
it’s not 100% sorting the viruses. but i do get significantly more reads in lower ranks.