We are trying to export the number of assigned reads for particular taxon to csv format, however, it seems that the output is always summarized counts. We can visually see the assigned numbers in the GUI, which differs from the summarized. The problem comes during the export, the numbers doesn’t match.
Procedure:
Highlight taxon level we wish to export
File -> Export -> csv format -> taxonPath_to_count (also tried taxonName_to_count) -> assigned -> tab -> filename
I am not sure why this is so and it seems to happen for both MEGAN5 (v5.11.3) and MEGAN6 (v6.4) versions.
If you request assigned counts, then MEGAN exports assigned counts, unless the node is collapsed, in which case the summarized count is exported. To avoid the latter, uncollapse any such node for which you don’t want this to kick in
Dear Daniel, I also have a question on this topic. in my original library the node that I am exporting there are sum=27355 and ass=12158, however when I open the extracted file (rma) it change to sum=12144 and ass=2571. if I export fast all the 27355 sequences are in the file? so what should I change to have all de 27355 sequences in the exported .rma file?
In the inspector window you can select some taxa and then use the File->Export Selected Taxa… menu item.
But probably the better way to export counts is to use the File->Export → Text (CSV) Format… menu item.
This allows you to select nodes in the taxonomy (or a functional) viewer and then to export counts, reads, etc in several different ways.
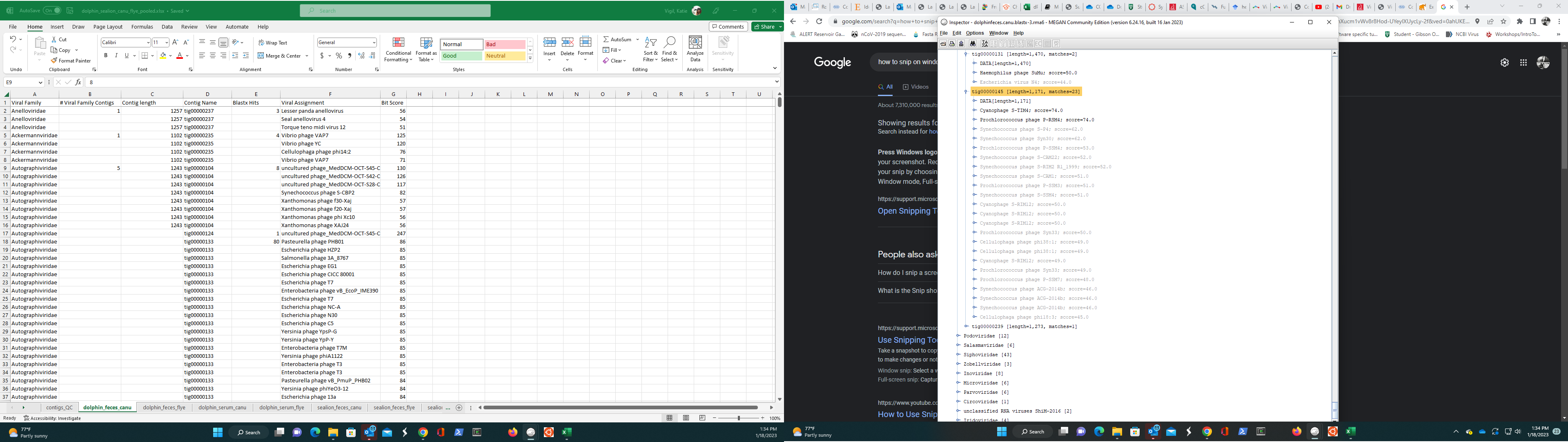
I was able to change the text file to a .csv file and I was able to extract select viral family FASTA files by right clicking the node on the tree and clicking “extract.” I was wondering now if there is a way to select a node and extract all the NCBI accession numbers? I only see the accession numbers listed on the inspector when you expand a node to see alignments. Is there a easier way to extract accession numbers? Thanks! Katie
This is where I see the accession numbers from the different nodes in the inspector window.
Query: 386 RANPRGTPSHQVVPHPLPTPGSFEVHVHNNCLCNEYLSLRNRVLQQVPEP-LDT-----FVDEMRNLAHRVSTWLGKHTPSDGEWIQQYSGRKATMYRNAAADLMLVPFSRRDRYIKSFL 45
R R TP Q P G ++V ++ +E +SLRNR+L +P+P L+T FV EMR+L H+V T + + I +Y+G K T Y AA LM P ++RD YI FL
Sbjct: 4 RDKTRATPWKQYCFKSFP–GWYKVDYPSSTYIDEEVSLRNRILLPMPQPQLNTPQWLSFVREMRHLKHQVPI—VETLTRQQVILKYTGAKRTRYEKAAISLMTKPLNKRDSYIDCFL 118
Query: 44 KPEKISDPT 18
K EK+ T
Sbjct: 119 KVEKMPHET 127
I have an update. Looks like I can’t export all the blastx alignments with e-value, % identity etc. from the inspector. However, I was able to export this information in .csv from highlighting all my viral families on the taxonomic tree. When you export from the inspector and change the .txt file to a .csv file all the data is still organized in one column making it hard to clean up for data analysis in R, so exporting from the taxonomic tree is the way to go. I am still in the process of learning how to visualize this same data using Diamond. I need to figure out the fastest way to create a table in excel with my virus family, genus & specie alignment, %identity, e-value, and protein aligned to my contigs, so I can create figures in R. Thanks!
Gray means that the bitscore for the alignment is more than 10% lower than the best bitscore for the read, and such alignments are not taken into account during analysis. This value is the “topPercent” threshold.
Hi Daniel,
Is there a way to export all a taxonomic information via command line version of MEGAN or Windows version that gives me a .tsv or .csv file similar to DIAMOND? Diamond produces a perfect .tsv table from all my blastx hits against the NCBI database, but did not provide order, subfamily, family, genus or species, so I ended up meganizing my blastx via Diamond into a .daa file and uploaded it to MEGAN, but I still can’t figure out how to generate the same table as Diamond with all the taxonomic information from the NCBI database. Can MEGAN do this? Below is an example