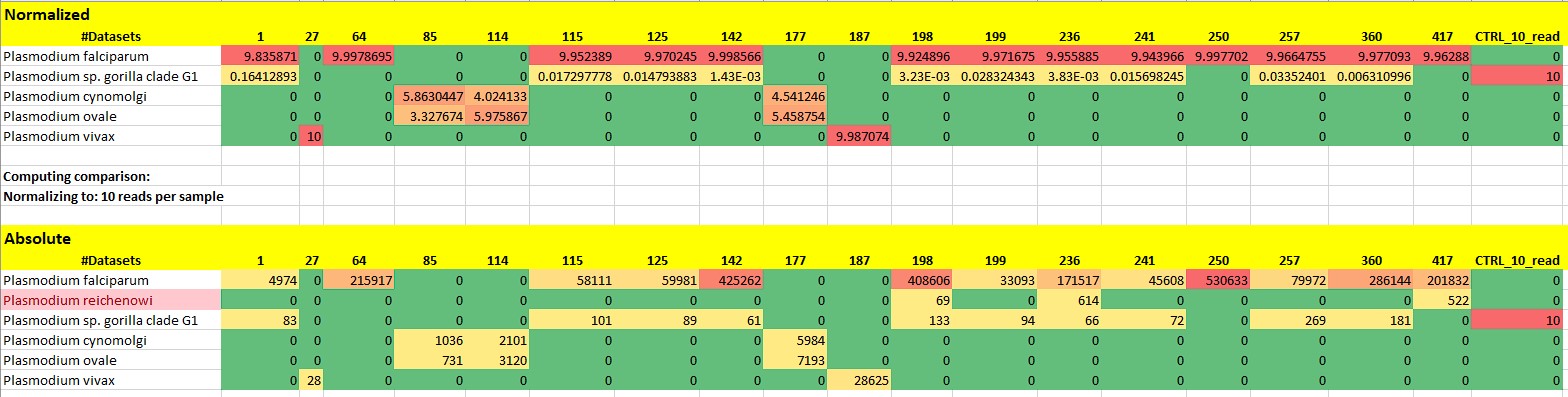
We were surprised to see a strange loss of data after normalization, despite using the “Keep at least 1 read” option (as in ignore all unnassigned reads checked). The figure below shows that the Plasmodium reichenowi taxon disappears completely, although it is stable presence with reads of 69, 614 and 522.
In both cases, a contig-based analysis was performed, abundance values were determined by remapping, and then the CSV import module was used to load the TaxID, reads values into Megan.
I was just about to make the same post, so I’m glad you made it blaize! I’ve found the same thing with my data in multiple projects.
Here are two examples using the same input files (same LCA parameters) with normalized and absolute comparisons (with “keep at least 1 read” checked). I assume this might be happening because the numbers become so small when one fasta has several orders of magnitude fewer reads, and the other files are forced to normalize down too much? But a workaround for this scenario would be much appreciated because sometimes I do want to normalize samples that have vastly different performances, and would like to reduce the problem of the inbuilt normalization mainly just making the good samples look far worse. Could something like the normalized file containing decimal proportions of the total reads be used so that it’s not comparing read counts anymore, but proportions? Just an idea.
Thanks for your confirmation! Yes, whatever is going on in the background when normalising, the output cannot be used to compare samples. For example in this case, the fact that the number of viral taxa is reduced by a fifth, when they are clearly there, is a serious issue. Summa summarum, until the next release arrives, external tools should be used to solve the normalisation.
What do you propose? The fact that you lose taxa through normalization could be seen as a feature, not a bug, because keeping all taxa in will make the samples with larger absolute counts look more species rich than the samples with smaller absolute counts.
I see two possibilities:
(re)-implement random subsampling to the smallest sample size. This was available in much older versions of MEGAN. (I took it out because the result is very similar to simply scaling down the counts, in particular, taxa with small counts still disappear…)
Use square-root scaling. I understand that this is considered a statistically sound approach. I could implement that.
By default, MEGAN scales nodes by square-root to indicate counts.
If the older random subsampling also removes taxa then we really have the same situation as now. If you think that could preserve the original taxon count using square-root scaling then we would really appreciate the implementation.
The point is not to change the taxon count in any way, for now we just want to see a little more balanced abundance landscape of the samples.
In any case, the current algorithm behaves strangely, because as I wrote above, the Plasmodium CTRL sample was preserved with only 10 reads, while P. reichenowi was removed, even though there are 69+614+522 reads.
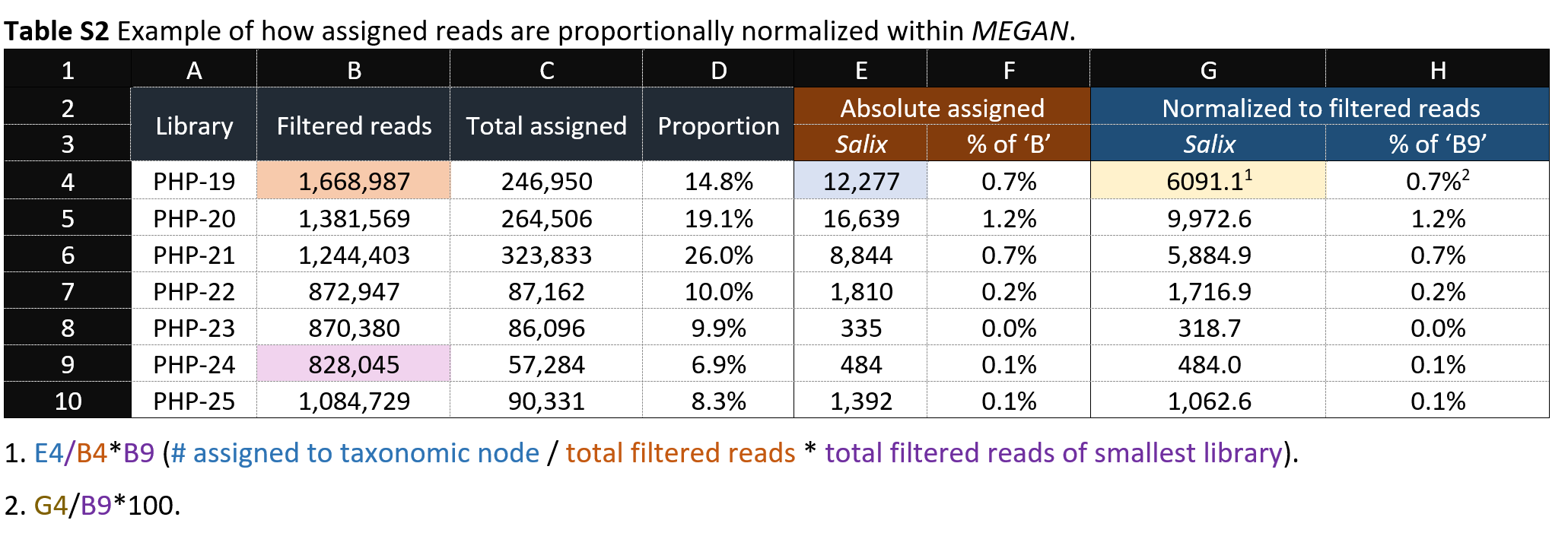
Would also a strategy where the normalized variant is converted from read counts to read percentages of each library work? So instead of comparing a transformed read count, it compares something like:
Sample 1, Taxon A 0.02%, S2 TA 0.0004%, S3 TA 1.3%, etc., where each percentage is derived simply by how many reads that taxon node makes up of the total reads for that library. So in the case of the table below, rather than converting the reads to be column G, converting them to be more like column F?
Perhaps a square-root normalization would be better in the end, but having a second normalization option (or in this case, a third) might be nice depending on the circumstances of the project. Sometimes a library tanks during read filtering (with ancient DNA in particular) where there are a lot of duplicates or something like that (a sample with poor DNA preservation). And so then one is stuck either removing that sample from the normalized comparison (even if it still has some informative reads) or including it but obscuring all the differences among the other samples because everything had to be normalized so far that many rare but informative organisms are dropped in the best-performing samples.
Is a strategy like that (the percentage conversion) reasonable within MEGAN?
Thanks, this percentage approach is also a very good idea, maybe I’ll try it on a real life dataset, but square root normalization would really be the way to go.
Also there’s an awful lot of material on the subject, here are some better ones: