I constructed some FASTQs with equal amount of reads from Escherichia coli and Pseudomonas aeruginosa, and assembled contigs using megahit. For the contigs binning I used the next commands:
One thing that I notice when looking at your file is that there appears to be an issue with the current version of DIAMOND.
For example, your sequence test_F0-N0-O0_479 has length 246 and received 3,103 alignments from DIAMOND. While many of the alignments are to different Pseudomonas species, these appear to have no affect because MEGAN uses a heuristic to avoid looking all all 3,103 alignments to place such a tiny contig.
Which version of DIAMOND did you use? Perhaps try using DIAMOND v0.9.36, available here:
Please let me know whether this solves the problem.
I have forwarded the issue to Benjamin Buchfink, the author of DIAMOND. He would like to have access to the original input file. Please send me an email and I’ll pass your email address on to him. You can find my email address here
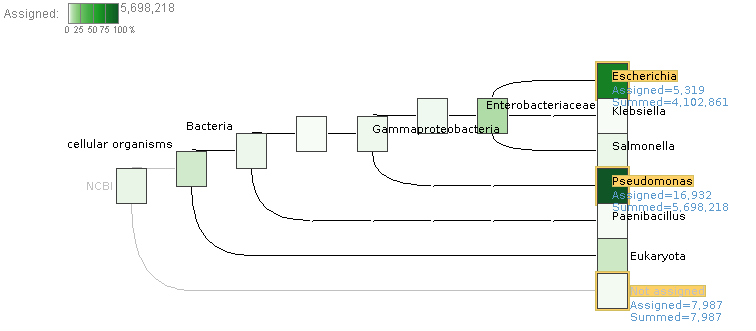
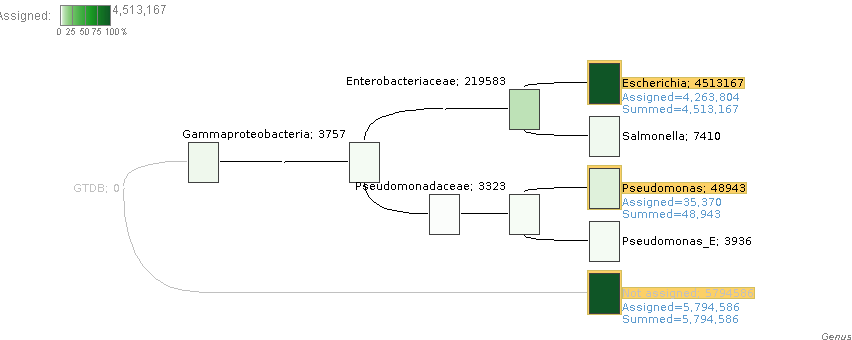
I have been testing and found something very odd, in test.daa I use the Inspect menu option in, for example, Pseudomonas aeruginosa B136-33. In there the sequence test_F0-N0-O0_155 has the next DATA:
but if i search this sequence in my fasta, only test_F0-N0-O0_1 (length: 1211306) has this sequence as a subsequence, and if I use diamond view on the test.daa file, test_F0-N0-O0_155 only has 25 matches and are all different than the ones I see in the inspect view.
Also, diamond view and the Inspect view reports a length of 2,655 for test_F0-N0-O0_155, but the actual length is 3049