I’m interested in removing/excluding entire genera from metagenomic analysis so I can look at relative differences of only a subset of nodes. Is there a way to do this in megan?
One way would be to output the read IDs of the undesired nodes and remove those from the original fastq, then rerun diamond, but I’m hoping this isn’t necessary…
You can setup taxa to be ignored:
To do so, open a new window and without loading any data, open and select ALL taxa that you want to disable.
Then, use the Edit->Preferences->Taxon disabling ->Disable… to disable all the selected taxa.
Alternatively, if you don’t select any nodes then you can paste in a list of NCBI taxon ids to ignore.
Unfortunately, this feature is not recursive so you do need to specify all taxa, from low to high ranks, that you want to ignore
You can select the nodes of interest and then use the “Extract to New Document” menu item to produce a new file that only contains the nodes of interest.
When using MEGAN’s charts, only select the nodes of interest.
If you want to use the Disable Taxa… mechanism that I previously described then please note that I have just updated it so that one can select a single node (e.g. Viruses) to disable all taxa on or below that node (e.g. disable all virus nodes).
Hi Daniel, thanks for the great software and support!
Is it possible to add multiple taxa to be disabled at one time using the Edit>Preferences>Taxon Disabling>Disable>Input window? Using a space, semi-colon or colon as a separator, results in “unknown taxon” error.
I have many species from diverse lineages that need to be disabled for each analysis. These are species hits from species I know do not occur in my study area (or country). For example I have a read that is assigned to Aves (Bird), but the included hits using my parameters are many hits from one species (Gallus gallus, chicken) and only one hit from New Zealand kiwi, which I am sure doesn’t occur in my European study area. This situation occurs for many species in many different animal lineages and greatly reduces the resolution of my results.
It is not possible to select multiple nodes in this case, because the node, for example, for NZ Kiwi is not shown. What I can do is make a manual list of all the species I wish to exclude, based on the included hits in the inspector window, and then add them to disabled taxa - but doing this one by one becomes prohibitive.
A great feature for this would be to have a way of adding species to the disabled taxa list from the inspector window (e.g. right click on hit and have an option like “add taxon to disabled taxa list”). That way, the manual lists I currently need to create (and subsequently get taxids for) could be done more rapidly.
entering multiple taxon ids or names separated by commas should work, I just tested it, seems fine.
I took a look at the code, a single unknown taxon will cause the command to abort. That is not ideal, so in the next release entering an unknown taxon will only cause a warning, other taxa will still be processed.
Another way to specify taxa to be disabled is to use the main window without any data. You can search for and select nodes in the tree viewer and then have them disabled. Note that if you disable an internal node then all nodes below will also be disabled.
I have also added a new context menu item to the Inspector window:
if you select one or more match nodes then you can have them disabled:
As usual, great response and fast updates. I have tested all three options and they both work perfectly as far as I can tell. The new addition to the context menu is a great help to me.
Hi! I have tried to disable some taxa from my tree using the Taxon disabling option… The taxa I wanted to remove still appears in the window. Does anyone know how I can fix this?
Hi, It´s working for me.
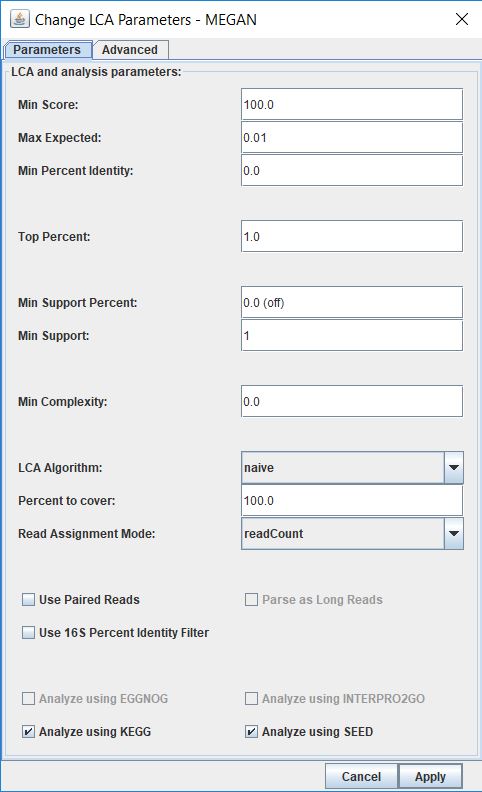
Once you disabled taxa, did you go to Options>Change LCA Parameters>Apply? You need to rerun the binning.
Are the taxa you wished to disable listed in Edit>Preferences>Taxon disabling>List Disabled?
Have you downloaded the latest build?
Which route are you taking to disable taxa - selecting on tree, inputting taxID, using context menu from visual inspector?
Bastian is right: the disabled taxon option is applied during data analysis.
The effect is that MEGAN will ignore any alignments to references associated with with any disabled taxon.
Hi Daniel, just wondering if it is still possible to disable a node, without disabling taxa below that node? Some sequences in genbank are labelled only to genus level, meaning that even reads with many hits to a single species will only get assigned at genus level, due to one poor genbank entry.
Thanks Daniel, I guess for now my work around will be to inspect results and reBLAST excluding problematic specific entries from the search database before re-importing to MEGAN.
If in the future there was an option of not applying the “mark all descendents as disabled” algorithm (e.g. perhaps using a symbol before individual taxids in the disable taxon dialogue), that would be great! (could also be a nice way in general to create versatile custom taxonomies).
Just a follow up question. Could you please explain why this BLAST hit only gets assigned at the genus level? The blast was done using NCBI nt database. The taxon C. aethiops sabaeus is in the NCBI taxonomy, as a synonym of Chlorocebus sabaeus. The organism specified for this particular entry is also Chlorocebus sabaeus.
Perhaps due to the min support parameter or min support percent parameter?
If a species does not attract enough reads then the reads are pushed up the taxonomy.
Does that explain it?
Sorry, I just took another look at your example:
The LCA is operating exactly as intended:
your read has similar-quality alignments to both Cercopithecus and
Chlorocebus and so the read is placed on their LCA, Cercopithecinae.
Hi Daniel, Thanks for your response again. That is true, the LCA is fine. What is confusing to me are a few particular BLAST hits and how they are being read from the blastXML by Megan before the LCA is applied.
Megan is reading the hits as “Cercopithecus” (genus level only), but these NCBI entries have species (or even subspecies) level tags. To simplify for an example, I have attached the results for blasting one sequence. Megan reads hit no. 5 as Cercopithecus (in inspector view), but the xml file def for this hit is Cecopithecus aethiops. Do you know why this might be?
I have attached the xml file also (megan file was too large).
So, the problem is that “Cercopithecus aethiops” is not in MEGANs current copy of the NCBI taxonomy, but it also looks like the current NCBI taxonomy doesn’t contain the species either…
Thanks for the detailed response Daniel. This is good to know. The workaround I used was to rerun the blasts specifically excluding the problematic NCBI entries (in this case just two GI numbers). Then all the reads end up in their expected taxon bins in megan. Thanks again.
I am adding this comment this because I recently had the same issue with a different species in a different data set. The issue was with NCBI synonyms. Including the nucl_acc2tax-Mar2018.abin file while importing the BLAST file fixed the issue.
Cercopithecus aethiops is listed in NCBI as one of the synonyms for Chlorocebus aethiops (the current NCBI genus name). My BLAST results only had “Cercopithecus aethiops”, so due to the absence of that species in the Megan-NCBI taxonomy, Megan assigned it to Cercopithecus.
Including the nucl_acc2tax-Mar2018.abin file while importing the BLAST file binned the hits to the current NCBI taxon used for this species (Chlorocebus aethiops). This also fixed a similar issue with another data set. At least for my needs, this means it probably makes sense to always include the nucl_acc2tax.abin when assigning taxonomy.